
Many interesting phenomena take place on the surface, one of which is adsorption. Adsorption is one of the key surface phenomena that has extensive practical applications, some of which include separation and purification of gases to obtain industrial gases of high purity [1], removal of organic and heavy metals pollutants from water [2], facilitation of catalytic reactions for production of industrial chemicals and pharmaceuticals [3], medicine and drug delivery [4], environmental remediation [5, 6], chromatography [7] and in preparation of surface films and coatings [8]. During the process of adsorption, the surface (adsorbent) attracts and holds the adsorbing molecule (adsorbate) by means of surface forces which may be physical or chemical in nature. Physical forces such as electrostatic interactions, ion-dipole attraction or van der Waals forces result in physisorptions which could be multi-layered and reversible whereas, chemical forces such as coordinate covalent bonding between some sites on the adsorbate and adsorbent results in monolayer chemisorption that is always difficult to reverse [9].
Adsorption also occurs on surfaces of metals and alloys, especially at their interface with some organic molecules that contain certain functional groups such as O-H, N-H, S-H, as well as multiple bonds, heterocyclic and aromatic rings [10]. This usually results in the formation of a surface adsorbed film that ‘blankets’ the surface from the medium within which it is in contact. For instance, surfaces of pipeline steel in contact with aggressive solutions such as acid, brines, bases, saline water, etc., are often protected from corrosion by introducing some organic molecules into the medium. The organic molecules differentially drift to the steel surface, become adsorbed on the steel active sites and form a protective film that shields the surface from corrosive attack [11]. This principle has been exploited in corrosion inhibition which finds application in various industries where metals and alloys regularly contact with various aggressive environments.
In assessing the extent of adsorption, the fractional coverage of the adsorbate on the adsorbent is often determined and analysed as a function of pressure or concentration for gaseous and liquid adsorbate systems, respectively. During the analyses of data, the associated parameters (surface coverage and concentration/pressure) may be used to construct adsorption isotherms based on suitable models. Detailed explanations on this has been provided in literature [9]. To obtain the surface coverage data, rigorous experiments is often conducted, which may be time consuming and expensive. The characterization of the deposited surface film, active adsorption sites to elucidate adsorption kinetics, energetics and mechanism may entail the deployment of several techniques such as FTIR, XPS and EDS. These are usually time and energy consuming and the associated equipment are very expensive and high energy demanding.
The use of computer, algorithms and software to model adsorption of various adsorbates on the surface of adsorbents provides an easier and less rigorous approach to examining adsorption phenomenon. With a workstation and appropriate software, it is easier and faster to model the adsorption of corrosion inhibitors, especially where there is dearth of research facilities and inadequate funding for experiments. The results are often accurate although quantitative data on corrosion inhibition efficiency is yet to be determined by computational approach.
In this study, computational intelligence is deployed to assess the formation of adsorbed species of organic molecules on steel surface. Since steel is composed mainly of iron with Fe (110) crystal surface, computational chemistry approach, including quantum chemical calculations and molecular dynamic simulations is deployed to predict the adsorption feasibility as well as the kinetics and energetics. This approach affords relative ease of understanding the adsorption phenomenon at less cost. Benzothiazole (BZT), and its four derivatives namely, benzothiazol-2-ol (BZT-OH), benzothiazol-2-amine (BZT-NH\(_2\)), benzothiazol-2-carboxylic acid (BZT-COOH) and benzothiazol-2-thiol (BZT-SH) were examined in the present study. With each compound containing the benzothiazole moiety and varying in attached substituents (Figure 1), the effects of the substituents on the adsorption behaviour and associated quantum parameters was the driving motivation behind the choice of the compounds.
Using experiments, it has already been demonstrated that some BZT based compounds have can efficiently adsorb on steel surface and inhibit its corrosion in acidic media [12, 13]. However, it is not clear how their structures correlate with the inhibition efficiencies. The active adsorption sites, mode of adsorption and the energetics associated with the binding of the molecules on the steel surface were not explained in these reports. The structural similarities were complex and not easy enough to observe slight differences. In other to provide such explanations and further insight into the structure-function relationship, effects of the substituents and possible active adsorption sites, the present work was designed and the results obtained from computational studies are hereby presented.
The computations were performed using Materials Studio (BIOVIA, Dassault Systèmes, 2017). Each molecule was built using the 3D Atomistic tool. The geometry of each molecule was optimized using the Forcite tool to obtain the most stable configuration and minimized energy [14].
Dmol\(^3\) was used to perform electronic structure modelling in the B3LYP functional, using the DNP basis set [14]. The HOMO (\(E_{\mathrm{HOMO}}\)) and LUMO (\(E_{\mathrm{LUMO}}\)) energies were obtained. Molecular reactivity indices were computed via: \[ \Delta E = E_{\mathrm{LUMO}} – E_{\mathrm{HOMO}}, \label{eq:gap}\ \tag{1}\] \[IE = -E_{\mathrm{HOMO}}, \label{eq:ie}\ \tag{2}\] \[EA = -E_{\mathrm{LUMO}}. \label{eq:ea} \tag{3}\]
Similarly, electronegativity (\(\chi\)), global hardness (\(\eta\)) and global softness (\(\sigma\)) were be calculated using Eqs. (4) – (6) [15]:
\[ \chi = \tfrac12(IE + EA), \label{eq:chi}\ \tag{4}\] \[\eta = \tfrac12(IE – EA), \label{eq:eta}\ \tag{5}\] \[\sigma = \eta^{-1}. \label{eq:sigma} \tag{6}\]
The Fukui electrophilic and nucleophilic functions were calculated by Mulliken population analysis using the optimized structure. Mulliken atomic charge distribution was calculated to screen the possible adsorption sites on the molecules [15].
Pure Fe metal structure was imported from Material Studio software and cleaved to a surface with a cleave plane of 1:1:0, resulting to surface vectors of U: 0.5 -0.5 0, V 0.5 -0.5 0. The cleaved surface was placed into three different layers and the symmetry was defined by creating a supercell with a range of 5\(\times\)5, where the adsorbate was placed 5Å from the surface [16]. A large vacuum of 15Å from the surface was created to avoid interaction of the adsorbate with both sides of the Fe surface due to boundary conditions. This resulted to a 3D lattice simulation box of 12.419 \(\times\) 12.419 \(\times\) 20.40. The Adsorption Locator tool was used for the simulation process [16]. The forcefield was set to the Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies, COMPASS. Location of the adsorption was set to the surface region defined by atom set with the adsorbate placed on the surface for annealing. Upon successful annealing, conformations of the adsorbate to the Fe (110) metal surface were obtained for each derivative. Also, the associated energy parameters such as total energy, absorption energy, deformation energy, rigid adsorption energy and activation energy were calculated [17].
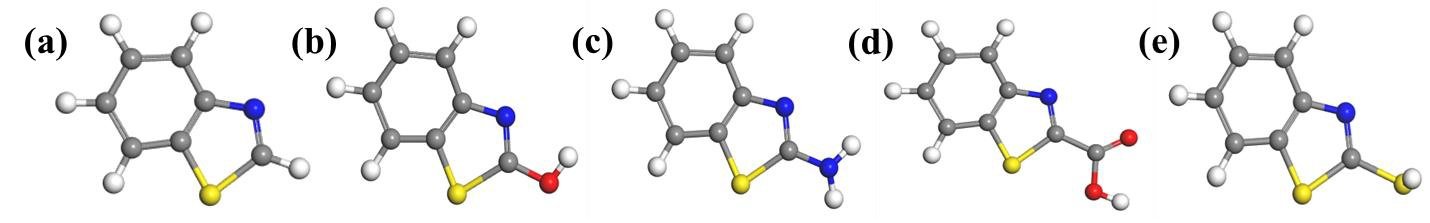
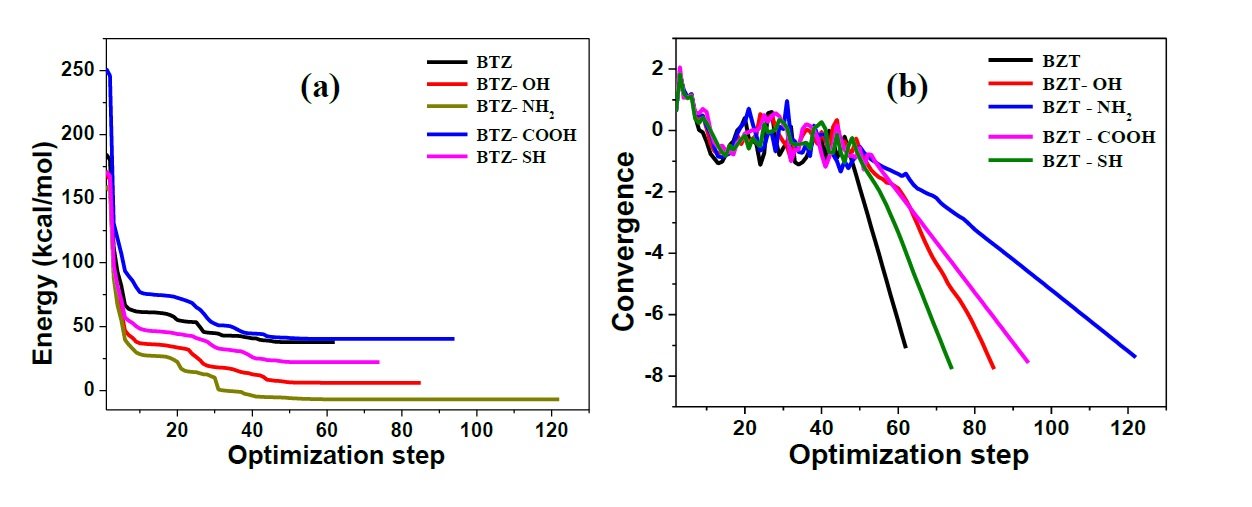
The geometry of BZT and the four derivatives studied were optimized to ground state minimum energy, and the optimized structures obtained are shown in Figure 2. Upon optimization, energy and convergence were obtained (Figure 3) indicating that a molecular configuration corresponding to the minimum energy has been reached. The optimized geometries appeared to be planar and this indicates that the compounds could adsorb efficiently to produce surface film on Fe. It has been established that planar geometrical molecules show better corrosion inhibition than less planar ones [18, 19]. Planar geometrical configuration enables molecules to cover a large area of the substrate surface, which in turn generates large surface coverage values than non-planar geometrical structures. Most or all of the planar molecules can be available to come in contact with metal surface, making adsorption more effective, and by implication, more efficient corrosion inhibition [20]. The planar configuration obtained from geometry optimization of the studied molecules thus provides prior insight that the molecules could likely adsorb on steel surfaces.
The energies of the frontier molecular orbitals, namely HOMO and LUMO, the band gap energy (\(\Delta E\)) and the total energy of each molecule was calculated and the obtained values are displayed in Table 1. All values were obtained in five (5) decimal places but approximated to and presented in four (4) decimal places. The HOMO energy (\(E_{\text{HOMO}}\)) of BZT was \(-0.2201\) Ha whereas the LUMO energy (\(E_{\text{LUMO}}\)) was \(-0.0283\) Ha. The values of \(E_{\text{HOMO}}\) obtained for BZT-OH, BZT-NH\(_2\), BZT-COOH and BZT-SH were \(-0.2172\) Ha, \(-0.1980\) Ha, \(-0.2330\) Ha and \(-0.2253\) Ha respectively. A close observation of the values reveals that when compared to the base molecule (BZT), the \(E_{\text{HOMO}}\) values increased in the presence of electron donating groups (EDGs), namely, –OH and –NH\(_2\), but decreased in the presence of electron withdrawing groups (EWGs), namely, –COOH and –SH. For \(E_{\text{LUMO}}\), the values for the molecules containing EDGs, namely, BZT-OH (\(-0.0171\) Ha) and BZT-NH\(_2\) (\(-0.0047\) Ha) also increased compared to that of BZT (\(-0.0283\) Ha), whereas the \(E_{\text{LUMO}}\) values for the molecules containing EWGs, namely, BZT-COOH (\(-0.0770\) Ha) and BZT-SH (\(-0.0450\) Ha) decreased compared to that of BZT.
| Parameters (Ha) | BZT | BZT-OH | BZT-NH\(_2\) | BZT-COOH | BZT-SH |
|---|---|---|---|---|---|
| \(E_{\mathrm{HOMO}}\) | -0.2201 | -0.2172 | -0.1980 | -0.2330 | -0.2253 |
| \(E_{\mathrm{LUMO}}\) | -0.0283 | -0.0171 | -0.0047 | -0.0770 | -0.0450 |
| \(\Delta E\) | 0.1918 | 0.2001 | 0.1933 | 0.1550 | 0.1803 |
| \(IE\) | 0.2201 | 0.2173 | 0.1980 | 0.2330 | 0.2253 |
| \(EA\) | 0.0283 | 0.0171 | 0.0047 | 0.0770 | 0.0450 |
| \(\chi\) | 0.1241 | 0.1172 | 0.1013 | 0.1550 | 0.1351 |
| \(\eta\) | 0.0959 | 0.1001 | 0.0967 | 0.7801 | 0.0902 |
| \(\sigma\) | 10.4254 | 9.9985 | 10.3461 | 1.2820 | 11.0913 |
Fundamentally, electron withdrawing groups (EWGs) reduce the electron density of a given molecule through the carbon atom to which they are attached, whereas electron donating groups (EDGs) increase the net electron density of a molecule. It can therefore be inferred from results that the presence of EWGs decrease LUMO and HOMO energy levels whereas the presence of EDGs increase LUMO and HOMO energy levels. This observation is in agreement with that reported by Trang and co-workers [21].
When applied to corrosion inhibition studies, the propensity of an adsorbate to attach to a metal surface by adsorption is driven more by the energy difference between the HOMO and LUMO, a parameter referred to as the energy gap (\(\Delta E\)), than by the individual energies of the frontier molecular orbitals. A smaller energy gap demonstrates good propensity for enhancement of the ability of the molecule to donate and accept electrons [22]. From Table 1, the magnitude of \(\Delta E\) follows the trend BZT-OH \(>\) BZT-NH\(_2\) \(>\) BZT \(>\) BZT-SH \(>\) BZT-COOH. Since the ease of adsorption is a function of lower energy gap, it therefore follows that electron donation and acceptance would be most enhanced in BZT-COOH and least enhanced in BZT-OH. In other words, adsorption of BZT-COOH could occur with most ease than the other molecules. However, the variation in the magnitudes of the energy gap values were not so significant, hence all the molecules may exhibit good reactivity and adsorption properties.
Molecular reactivity is also expressed by other indices such as ionization energy (IE), electron affinity, electronegativity, and global hardness and softness. These parameters are closely related to and are often estimated from the values of both \(E_{HOMO}\) and \(E_{LUMO}\). The values of these parameters were calculated and also displayed in Table 1.
The value of IE, also known as the ionization potential, is directly related to \(E_{\text{HOMO}}\) and [in this context] represents the least amount of energy needed for electrons to attach to the surface of the metal to form a bond or adsorbed layer. In other words, low ionization potential signifies high ease of release of valence electron to the metal surface, leading to increased adsorptive interactions [23]. As can be seen in Table 1, BZT-NH\(_2\) exhibited the lowest IE, followed by BZT-OH whereas BZT-COOH exhibited the highest ionization energy. Compared to BZT, the –NH\(_2\) and –OH substituent reduced the ionization energy whereas the –COOH and –SH substituents increased the ionization energy. Therefore, the ease of electron release and adsorptive interaction would follow the trend BZT-NH\(_2\) \(>\) BZT-OH \(>\) BZT \(>\) BZT-SH \(>\) BZT-COOH, and that adsorption of BTZ-NH\(_2\) would be associated with more ease, followed by BZT-OH and would occur via electron donation to the metal orbital [24]. This inference is consistent with classification of the –NH\(_2\) and –OH groups as EDGs.
On the other hand, the energy released when an atom receives an electron from metallic orbitals is expressed using the magnitude of a parameter called electron affinity (EA). The EA is therefore closely associated with the \(E_{\text{LUMO}}\) and higher EA signifies higher propensity to receive back-donated electrons from the metal orbital. It can also be observed from Table 1 that EA is highest with BZT-COOH (0.0770 Ha), followed by BZT-SH (0.0450 Ha) and lowest (0.0171 Ha) with BZT-NH\(_2\) (0.0047 Ha) followed by BZT-OH (0.0171 Ha). Thus, the adsorption of both BZT-COOH and BZT-SH would be facilitated more by back-donation of electrons from the metal to the molecules.
Global hardness (\(\eta\)) and global softness (\(\sigma\)) are additional quantum chemical parameters that may be used to predict molecular reactivity and also compare the properties of surface adsorbed molecules. The softer a molecule, the more reactive it is considered than the harder counterparts [25]. Results obtained from quantum calculations reveal that decreasing order of global hardness is BZT-COOH \(>\) BZT-OH \(>\) BZT-NH\(_2\) \(>\) BZT \(>\) BZT-SH whereas the global softness follows the reverse trend. This trend indicates that BTZ-COOH exhibits the highest hardness (or lowest softness) whereas BZT-SH exhibits the lowest hardness (or highest softness). It can be implied that the presence of –SH substituent conveys more softness (or high reactivity) on the benzothiazole moiety, followed by –NH\(_2\) group, then –OH group.
Electronegativity is another quantum parameter that can be used to comparatively predict the adsorption behaviour of molecules on metal surfaces. The impact of electronegativity values spans across propensity for charge transfer, adsorption energy, surface bonding, molecular orientation and competitive adsorption. For instance, the presence of highly electronegative atoms may enhance strength of the molecule–metal interaction, increase adsorption energy and enhance adsorption. In terms of orientation, the electronegative atoms may point towards the surface, maximizing interactions. Highly electronegative species on the adsorbing molecule could form coordinate covalent bonds or electrostatic interactions with species on the metal surface. Also, molecules with higher electronegativity values could outcompete others at surface sites, leading to improved surface coverage. The impact of electronegativity depends on the surface properties (e.g. roughness, Fermi level) of the metal, molecule–surface distance, molecular shape and size, temperature and pressure, and presence and concentration of other adsorbate species.
According to the electronegativity equalization theory, molecules with overall lower electronegativity values are more effective at inhibiting corrosion. Lower electronegativity allows these molecules to exhibit a larger electronegativity difference compared to the metal surface, making them better at forming protective layers or adsorbing onto the metal surface. The electronegativity value of iron is 1.74, 1.83 and 1.91 on Mulliken-Jaffé, Pauling and Allred-Rochow scales respectively. However, the Pauling scale is the commonly used scale which assigns Fe an electronegativity value of 1.83. The larger the difference between 1.83 and the electronegativity value of a given molecule, the stronger the adsorption strength. Results from present study reveals that BZT-NH\(_2\) exhibits the lowest electronegativity value, followed by BZT-OH whereas BZT-COOH exhibits the highest followed by BZT-SH. This implies that BZT-NH\(_2\) has the largest electronegativity difference with Fe, and may exhibit strongest adsorptive interaction, followed by BZT-OH. The BZT-COOH afforded the highest electronegativity value, and may possibly show less corrosion inhibition efficiency than the other molecules. The above inference is consistent with theoretical electronegativity order considering that the electronegativity value of O (hydroxy), N (amine), S (mercapto) and C (carboxyl) is 3.44, 3.04, 2.85 and 2.5 respectively. Thus, while BZT-NH\(_2\) may likely form stronger bonds, BZT-OH may be nucleophilic and basic whereas BZT-SH may be nucleophilic and less acidic.
The adsorption of organic molecules on surfaces of metals and alloys, such as steel, occurs through donor-acceptor interactions that take place at electron rich centres or heteroatoms. Local reactivity analyses often afford salient information on the adsorption sites of the inhibitor. To locate these sites, Fukui functions, Mulliken atomic charge distribution, and frontier molecular orbital plots were determined.
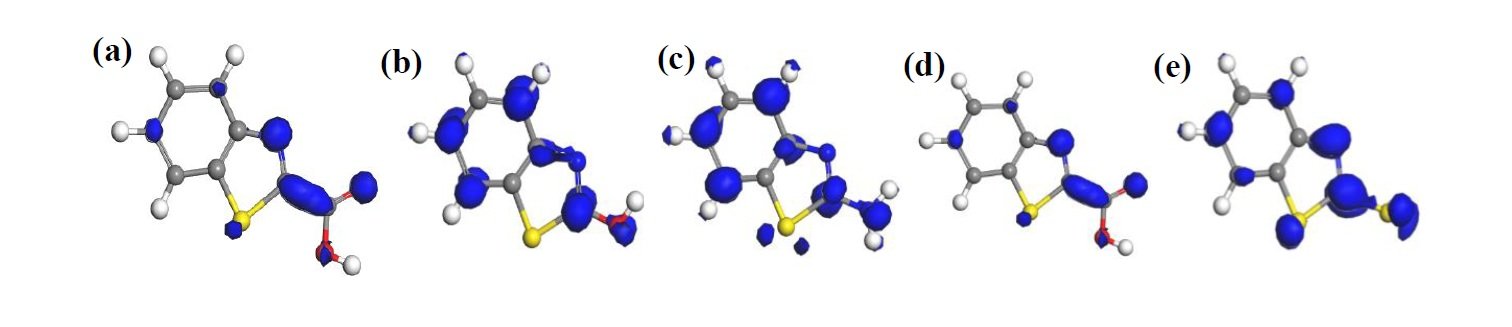
The HOMO and LUMO diagrams obtained for the studied compounds are displayed in Figure 4 and Figure 5, respectively. The HOMO orbital represents the space within where electrons are available for donation to acceptor species. Conversely, the LUMO orbital represents where electrons are unavailable and high propensity for the orbital to accept electron from donating species. From Figure 4, it can be observed that HOMO orbitals of all the compounds are mainly distributed over their entire aromatic ring indicating that the ring in molecules is potential active sites for electrophilic attack by the metallic cations. The molecules exhibit pi-character delocalized over most of the atomic rings. Thus, during adsorptive interaction with steel surface, the delocalised HOMO electron densities would favour forward donation of electrons to vacant d-orbitals of Fe.
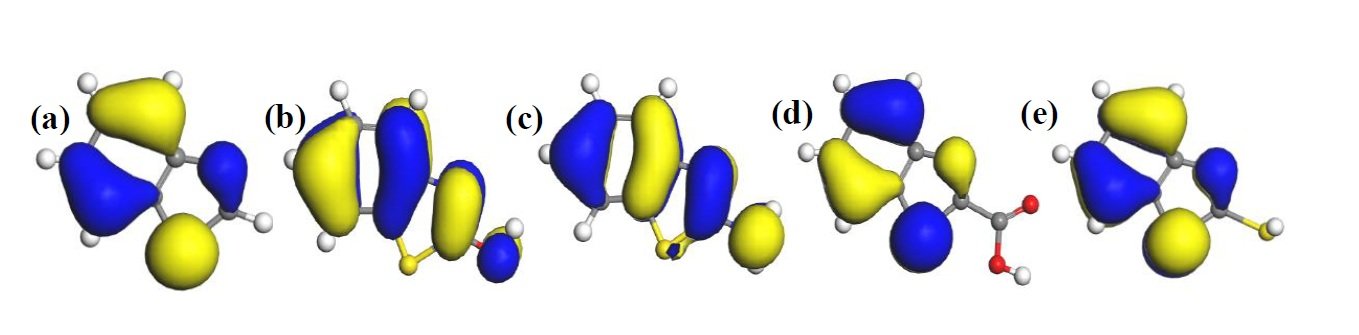
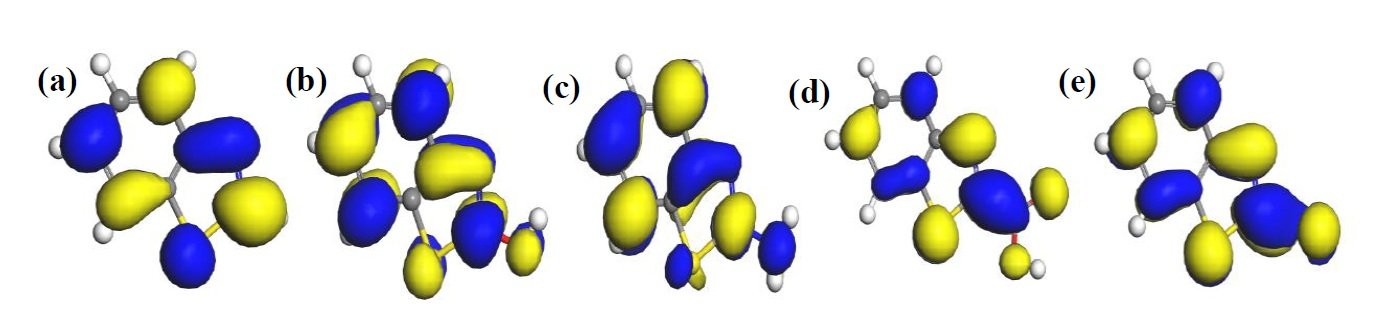
The presence of different attached groups on the BZT backbone also conveys additional but different potential active sites on the molecules. For instance, additional HOMO orbitals were observed around N heteroatom and the adjacent double bond for BZT-OH and BZT-NH\(_2\). Also, the O and N atoms in the hydroxyl and amino groups in BZT-OH and BZT-NH\(_2\) respectively, were found to be sites with high HOMO electron density whereas the S and COO atoms in BZT-SH and BZT-COOH were not. Rather, instead of HOMO at the around N heteroatom and the adjacent double bond, the HOMO was found around the S heteroatom. Thus, while N heteroatom is a potential adsorption site for BZT-OH and BZT-NH\(_2\), S heteroatom is the potential active site for BZT-COOH and BZT-SH. On the other hand, it can be observed from Figure 5 that the LUMO diagrams are distinctly different than the HOMO diagrams. For all the molecules, the LUMO orbitals are mainly distributed around the aromatic C centres, N and S heteroatoms and the C atom bearing the attached group. Additional LUMO sites can be observed at O, N, S and COO of the attached groups on BZT-OH, BZT-NH\(_2\), BZT-SH and BZT-COOH respectively. These locations are potential sites with disposition to accept electrons from metal species in back-donation adsorptive interactions.
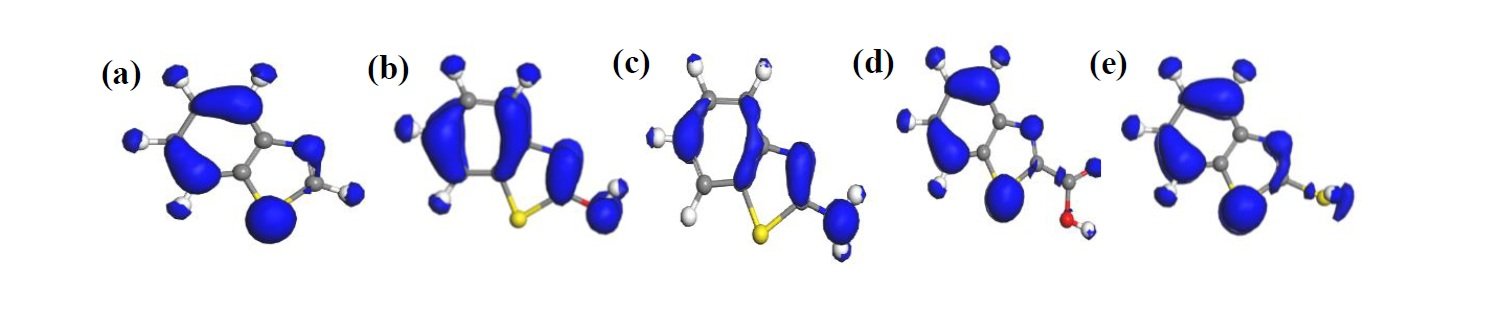
Fukui’s function is often deployed to describe chemical reactivity, especially potential sites where nucleophilic or electrophilic attack could take place in molecules [26]. Occurrence of nucleophilic and electrophilic attack is most likely to be associated with sites where the \(f^+\) and \(f^-\) value, respectively, is maximum. The larger the \(f^+\) and \(f^-\) value, the more reactive that potential active site may be [24, 26]. In other words, the most preferable site for nucleophilic attack or accepting electron from metallic species exhibits the highest \(f^+\) value, whereas the most preferable site for electrophilic attack or electron donation to the vacant orbital of Fe exhibits the highest \(f^-\) value.
In Tables 2 and 3, where the results obtained for Fukui indices for nucleophilic attack and electrophilic attack, respectively, are displayed, it can be seen that the most reactive site for both nucleophilic and electrophilic attack in all the molecules is the S(9) atom. The S(9) atom shows overriding reactivity in benzothiazole and all the derivatives, making it most likely to form a back bond with Fe(110) surface species through accepting electrons. The S(9) atom also exhibits predisposition to donate electrons to an open d-orbital on the surface of Fe(110) so as to create a coordinate bond. This and other potential sites are presented in bold in Tables 2 and 3 and also displayed in the Fukui diagrams (Figures 6 and 7).
| BZT | BZT-OH | BZT-NH\(_2\) | BZT-COOH | BZT-SH | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Atom | \(f^+\) | Atom | \(f^+\) | Atom | \(f^+\) | Atom | \(f^+\) | Atom | \(f^+\) | |
| C1 | 0.065 | C1 | 0.067 | C1 | 0.054 | C1 | 0.045 | C1 | 0.055 | |
| C2 | 0.046 | C2 | 0.078 | C2 | 0.087 | C2 | 0.015 | C2 | 0.028 | |
| C3 | -0.001 | C3 | -0.011 | C3 | -0.012 | C3 | 0.011 | C3 | 0.005 | |
| C4 | 0.020 | C4 | 0.033 | C4 | 0.032 | C4 | -0.004 | C4 | 0.007 | |
| C5 | 0.083 | C5 | 0.103 | C5 | 0.105 | C5 | 0.049 | C5 | 0.065 | |
| C6 | 0.000 | C6 | -0.003 | C6 | -0.001 | C6 | 0.008 | C6 | 0.003 | |
| N7 | 0.083 | N7 | 0.006 | N7 | 0.047 | N7 | 0.110 | N7 | 0.099 | |
| C8 | 0.136 | C8 | 0.101 | C8 | 0.060 | C8 | 0.060 | C8 | 0.079 | |
| S9 | 0.176 | S9 | 0.150 | S9 | 0.164 | S9 | 0.169 | S9 | 0.173 | |
| H10 | 0.081 | H10 | 0.070 | N10 | 0.040 | C10 | 0.087 | S10 | 0.144 | |
| H11 | 0.069 | H11 | 0.085 | H11 | 0.084 | O11 | 0.127 | H11 | 0.072 | |
| H12 | 0.076 | H12 | 0.076 | H12 | 0.082 | O12 | 0.045 | H12 | 0.059 | |
| H13 | 0.075 | H13 | 0.081 | H13 | 0.083 | H13 | 0.044 | H13 | 0.065 | |
| H14 | 0.090 | H14 | 0.079 | H14 | 0.079 | H14 | 0.065 | H14 | 0.067 | |
| H15 | 0.034 | H15 | 0.056 | H15 | 0.051 | H15 | 0.078 | |||
| H16 | 0.040 | H16 | 0.056 | |||||||
| H17 | 0.061 | |||||||||
| BZT | BZT-OH | BZT-NH\(_2\) | BZT-COOH | BZT-SH | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Atom | \(f^-\) | Atom | \(f^-\) | Atom | \(f^-\) | Atom | \(f^-\) | Atom | \(f^-\) | |
| C1 | 0.032 | C1 | 0.080 | C1 | 0.068 | C1 | 0.028 | C1 | 0.029 | |
| C2 | 0.084 | C2 | 0.045 | C2 | 0.024 | C2 | 0.084 | C2 | 0.085 | |
| C3 | -0.011 | C3 | 0.007 | C3 | 0.018 | C3 | -0.011 | C3 | -0.011 | |
| C4 | 0.015 | C4 | 0.042 | C4 | 0.029 | C4 | 0.013 | C4 | 0.015 | |
| C5 | 0.093 | C5 | 0.063 | C5 | 0.047 | C5 | 0.090 | C5 | 0.090 | |
| C6 | 0.032 | C6 | 0.011 | C6 | 0.017 | C6 | 0.035 | C6 | 0.033 | |
| N7 | 0.080 | N7 | 0.091 | N7 | 0.099 | N7 | 0.074 | N7 | 0.076 | |
| C8 | 0.025 | C8 | 0.054 | C8 | 0.035 | C8 | 0.024 | C8 | 0.002 | |
| S9 | 0.239 | S9 | 0.147 | S9 | 0.149 | S9 | 0.227 | S9 | 0.226 | |
| H10 | 0.084 | H10 | 0.099 | N10 | 0.113 | C10 | 0.018 | S10 | 0.097 | |
| H11 | 0.083 | H11 | 0.093 | H11 | 0.085 | O11 | 0.062 | H11 | 0.080 | |
| H12 | 0.087 | H12 | 0.074 | H12 | 0.066 | O12 | 0.005 | H12 | 0.080 | |
| H13 | 0.084 | H13 | 0.080 | H13 | 0.072 | H13 | 0.030 | H13 | 0.084 | |
| H14 | 0.072 | H14 | 0.097 | H14 | 0.076 | H14 | 0.079 | H14 | 0.081 | |
| H15 | 0.034 | H15 | 0.053 | H15 | 0.079 | H15 | 0.033 | |||
| H16 | 0.050 | H16 | 0.082 | |||||||
| H17 | 0.081 | |||||||||
The distribution of Mulliken atomic charges as presented in Table 4 demonstrates that for BTZ molecules, C(4) is the most positive center whereas N(7) is the most negative center followed by S(9). For both -OH and -NH\(_2\) derivatives, C(8) is the most positive center whereas O(10) is the most negative center followed by N(7). For the -COOH derivative, the most positive center is located at C(10) followed by O(12) whereas the most negative center is located at N(7). Also, for -SH derivative, C(4) is the most positive center whereas N(7) is the most positive center followed by S(10). In other words, for all the molecules, N(7) is a negative center and potential electron rich site and would participate in adsorption via electron donation. The interactions between these positive and negative sites of the organic molecules and Fe surface species could be electrostatic in nature or involving formation of coordinate covalent bonds, potentially leading to physisorption or chemisorption of the molecules on the Fe(110) surface [15].
| BZT | BZT-OH | BZT-NH\(_2\) | BZT-COOH | BZT-SH | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Atom | q | Atom | q | Atom | q | Atom | q | Atom | q | |
| C1 | -0.111 | C1 | -0.117 | C1 | -0.106 | C1 | -0.104 | C1 | -0.101 | |
| C2 | -0.115 | C2 | -0.124 | C2 | -0.117 | C2 | -0.114 | C2 | -0.105 | |
| C3 | 0.167 | C3 | 0.162 | C3 | 0.163 | C3 | 0.159 | C3 | 0.163 | |
| C4 | 0.241 | C4 | 0.248 | C4 | 0.254 | C4 | 0.238 | C4 | 0.243 | |
| C5 | -0.128 | C5 | -0.136 | C5 | 0.130 | C5 | -0.121 | C5 | -0.101 | |
| C6 | -0.093 | C6 | -0.091 | C6 | -0.090 | C6 | 0.093 | C6 | -0.088 | |
| N7 | -0.313 | N7 | -0.384 | N7 | -0.372 | N7 | -0.286 | N7 | -0.260 | |
| C8 | 0.089 | C8 | 0.435 | C8 | 0.360 | C8 | 0.101 | C8 | 0.267 | |
| S9 | -0.233 | S9 | -0.219 | S9 | -0.227 | S9 | -0.155 | S9 | -0.159 | |
| H10 | 0.072 | O10 | -0.441 | N10 | -0.379 | C10 | 0.554 | S10 | -0.258 | |
| H11 | 0.083 | H11 | 0.070 | H11 | 0.082 | O11 | 0.439 | H11 | 0.090 | |
| H12 | 0.081 | H12 | 0.082 | H12 | 0.091 | O12 | 0.454 | H12 | 0.100 | |
| H13 | 0.075 | H13 | 0.077 | H13 | 0.088 | H13 | 0.279 | H13 | 0.099 | |
| H14 | 0.086 | H14 | 0.073 | H14 | 0.084 | H14 | 0.080 | H14 | 0.092 | |
| H15 | 0.267 | H15 | 0.192 | H15 | 0.090 | H15 | 0.188 | |||
| H16 | 0.206 | H16 | 0.092 | |||||||
| H17 | 0.083 | |||||||||
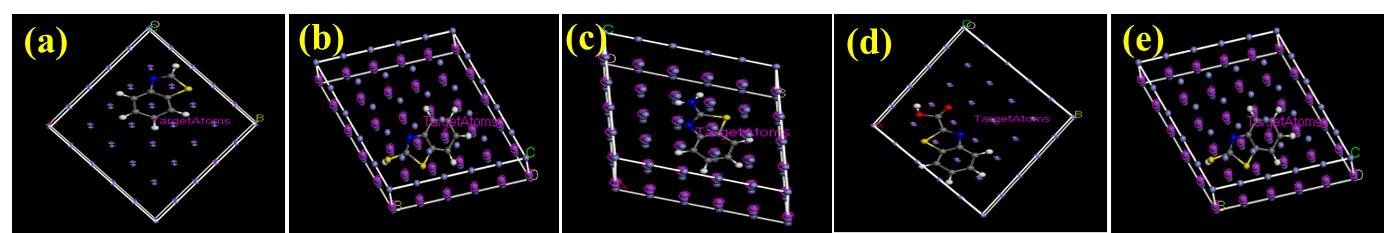
The mode of interaction of an adsorbate on the adsorbent surface can be modelled by MD simulations. The simulation is has used accurately predict and explain the possible type of molecular configuration of organic corrosion inhibitors on steel surfaces [14, 27]. In solution, an inhibitor molecule has to initially migrate to the substrate surface in order to adsorb effectively, then dislodge initially adsorbed water molecules [9]. The effectiveness of the adsorption and the consequential corrosion inhibition effect depends on how the molecule is oriented on the surface. Therefore, configurations of each of the studied molecules on the Fe(110) surface were scrutinized and the obtained results are shown in Figure 8 and 9. Results reveal that the adsorption of the molecules are theoretically feasible. The molecules were oriented on the surface in a planar configuration which indicates effective adsorption. Effective adsorption would result in good corrosion inhibition effect [29]. The associated energies obtained are displayed in Table 5.
| Parameter | BZT | BZT-OH | BZT-NH\(_2\) | BZT-COOH | BZT-SH |
|---|---|---|---|---|---|
| Total Energy | -23.7793 | -0.2172 | -76.2515 | -39.7265 | -50.7989 |
| Adsorption Energy | -61.6145 | -0.0171 | -69.3239 | -291.0945 | -73.0346 |
| Binding Energy | -38.3577 | -42.7607 | -41.6854 | -50.0221 | -53.1029 |
The binding energy of a given molecule is an important indicator of strong or weak adsorption as well as how effective the adsorption would be. The binding energy quantifies the strength of the interaction between a molecule (inhibitor) and the metal surface. Theoretically, molecules that exhibit stronger (more negative) binging energies adsorb more strongly and consequently shows better corrosion inhibition [17]. Therefore, the benzothiazole derivative with the strongest binding energy is expected to bind more effectively on Fe (110) surface and efficiently block the active sites where corrosion would have occurred on the surface. The consequence of this is the creation of a barricade that precludes corrosive species (like water, chloride ions, etc.) from reaching the metal surface and initiating corrosion.
Based on the results obtained (Table 5), the BTZ-SH molecule exhibits the most negative (strongest) binding energy ad is thus expected to afford the highest corrosion inhibition effect, followed by BTZ-COOH, BTZ-NH\(_2\), BTZ-OH, and BTZ. Based on this observation, it can be inferred that as expected, the attached groups on the BTZ moiety contribute to improving the inhibition efficiency of the base molecule, and the contribution will follow the trend -SH \(>\) – COOH \(>\) -NH\(_2\) \(>\) -OH.
It has been inferred from quantum molecular calculations that BTZ-SH shows the highest global softness index among all the studied molecules, indicating that it would exhibit the highest reactivity. This prediction is further buttressed by result of the MD simulations which shows that BTZ-SH has the highest binding energy among the five molecules. Being the softest, and potentially the most reactive, BTZ-SH also binds most effectively on Fe(110) surface, and would likely afford the highest inhibition efficiency if subjected to experiment. The binding energy of the studied molecules, and in extension, their strength of adsorption on Fe(110) surface, is thus largely dependent on the specific functional group since the crystallographic orientation of iron species in the steel surface does not change.
The strength of the interaction between an adsorbate molecule (inhibitor) and the adsorbent surface (Fe (110) is predicted using the quantified using the adsorption energy. A more negative adsorption energy value connotes a stronger binding interaction. Molecules that exhibit higher adsorption energy will be more strongly adsorbed or more tightly bound to the metal surface and prevent corrosive species from reaching the surface to initiate corrosion. From the results displayed in Table 5, BTZ-COOH showed the highest adsorption energy, followed by BTZ-SH, then BTZ-NH\(_2\) and BTZ. The adsorption energy of BTZ-OH was very low compared to the other molecules. Although the trend of adsorption energy is dissimilar to that of binding energy (which does not necessarily need to be), the -COOH derivative was initially found to exhibit the highest ionization energy of all the studied molecules. In addition, it was already inferred from quantum calculations that the -COOH derivative afforded the least band gap energy and that adsorption is most enhanced in BTZ-COOH. These observations are in agreement and further buttresses the results of MD simulations.
Corrosion inhibition often occurs by either adsorption or film forming mechanism whereby an organic molecule binds to the surface of the steel and forms a thin protective layer that barricades the surface from the corroding species [11]. The adsorption may be driven by physical or chemical forces, described as physisorption or chemisorption mechanism, respectively. The nature of the binding mechanism may be predicted from electronic interactions (such as charge density and electron transfer), frontier molecular orbital energy gap, the magnitude of the adsorption energy, bond angle analyses, adsorption dynamics and adsorption energy. Adsorption energy is the most reliable and a sufficient of all because it gives a quantitative data. Typically, very highly negative adsorption energies above -80 kJ/mol are indicative of chemical adsorption whereas adsorption energies less negative than -40 kJ/mol indicate physical adsorption mechanism [29, 30] Results displayed in Table 5 show that when converted to kJ/mol, all the adsorption energies are more negative than -80 kJ/mol, hence the various derivatives would adsorb by chemisorption. Chemisorption involves donation of electrons to the metal surface and it is usually stronger than physical adsorption, further enhancing the protective film [31].
The adsorption of some benzothiazole-based compounds, namely, benzothiazol-2-ol (BZT-OH), benzothiazol-2-amine (BZT-NH\(_2\)), benzothiazol-2-carboxylic acid (BZT-COOH) and benzothiazol-2-thiol (BZT-SH) on Fe(110) surface was theoretically assessed using quantum computations and molecular dynamics simulations within the DFT domain. The various parameters assessed were compared to the BTZ molecule to understand the effect of the substituent group on the reactivity. Compared to Frontier molecular orbitals energy, EL and EH values increased in the presence of electron with drawing groups (namely, -NH\(_2\) and -OH) and decreased in the presence of electron donating groups (namely, -COOH and -SH). The band gap energy decreased according to the trend BTZ-OH > -NH\(_2\) > BTZ > -SH > -COOH indicating that the ease of adsorption could be highest with BTZ-OH than all the molecules and as well least with BTZ-COOH. BTZ-OH is also the hardest molecule among the five whereas BTZ-SH is the softest, hence BTZ-SH could exhibit the highest reactivity while BTZ-OH could exhibit the least reactivity. The active site for both nucleophillic and electrophilic attack in all the molecules is S(9) atom and this may be the major adsorption site, followed by N and O atoms. On adsorption, the molecules assume a planar configuration indicative of efficient adsorption. Adsorption of the molecules would occur through chemical bonds or electron donation-acceptance mode (chemisorption) with BTZ-COOH and BTZ-SH exhibiting the highest adsorption and binding energy, respectively.